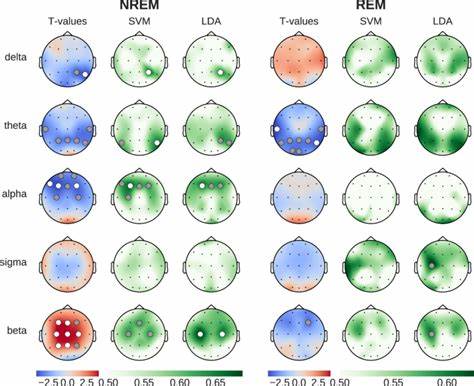
Huntington-Krankheit (HD) zählt zu den erblichen neurodegenerativen Erkrankungen, deren Ursache in der Expansion trinukleotidischer Wiederholungen im HTT-Gen liegt. Diese Wiederholungen, bestehend aus der Abfolge CAG, kodieren eine verlängerte Glutaminsequenz im Huntingtin-Protein, deren Länge eng mit dem Erkrankungsbeginn und dem Schweregrad der Symptome korreliert. Je länger diese Repeat-Region bei Geburt ist, desto früher treten die Symptome auf und desto schneller schreitet die Krankheit fort. Eine der großen Herausforderungen bei HD ist die sogenannte somatische Instabilität der CAG-Trakte, bei der sich die Anzahl der Wiederholungen im Verlauf des Lebens in verschiedenen Geweben – besonders im Zentralnervensystem – weiter vergrößert. Diese somatischen Erweiterungen tragen maßgeblich zum Fortschritt der Neurodegeneration bei und sind daher ein attraktives Ziel für innovative Therapieansätze.
In den letzten Jahren haben Wissenschaftler intensiv daran gearbeitet, Wege zu finden, die somatische Expansion dieser CAG-Tripletts aufzuhalten oder sogar rückgängig zu machen. Vor kurzem hat die Entwicklung von Basen-Editing-Technologien, einer weiterentwickelten Form der CRISPR-Cas9-Genom-Editierung, neue Perspektiven eröffnet. Im Gegensatz zur klassischen CRISPR-Methode, die DNA-Doppelstrangbrüche induziert, verändert Basen-Editing einzelne Nukleotide gezielt, ohne die DNA-Stränge zu durchtrennen. Diese präzise Methode minimiert potenzielle unerwünschte Mutationen und Zellschädigungen. Forschungen aus 2025 haben erstmalig gezeigt, dass das Einführen gezielter Unterbrechungen innerhalb des CAG-Repeats durch Basen-Editing in patienteneigenen Zellen sowie in Mausmodellen zu einer signifikanten Reduktion der somatischen Erweiterungen führt.
Dabei werden bestimmte Cytosin- und Adeninbasen gezielt modifiziert, um natürliche, nicht-pathogene Sequenzvarianten nachzubilden, die bei Menschen mit stabileren oder milderen Krankheitsverläufen beobachtet werden. Konkret bedeutet dies, dass Teile der CAG-Sequenz in CAA umgewandelt werden, welche zwar die Glutaminsequenz nicht verändert, aber die Instabilität der Wiederholungen deutlich mindert. Die praktischen Untersuchungen erfolgten in menschlichen Fibroblasten von HD-Patienten sowie in Htt.Q111-Mäusen, einem etablierten Huntington-Tiermodell mit verlängerten CAG-Repeat-Strecken. Mit Hilfe der Adeno-assoziierten Virus (AAV) Vektortechnologie wurde das Basen-Editiersystem in die zentralen Hirnregionen injiziert.
Resultat war eine dauerhafte und effiziente Unterbrechung der CAG-Region in bis zu 70 Prozent der Zellen, was eine messbare Verlangsamung und teilweise Umkehr der CAG-Erweiterung bewirkte. Die Anzahl der Wiederholungen nahm um mehrere Einheiten ab, wodurch eine potentielle therapeutische Schwelle unterschritten wurde, die mit reduziertem neurodegenerativem Schaden korreliert. Diese Erkenntnisse bauen auf frühere epidemiologische Studien auf, welche zeigten, dass Patienten, deren CAG-Repeats natürliche Unterbrechungen wie CAA enthalten, einen späteren Erkrankungsbeginn und eine verzögerte Krankheitsprogression aufweisen. Die nun mögliche künstliche Einfügung solcher Unterbrechungen lässt auf einen direkten kausalen Zusammenhang schließen und stellt eine vielversprechende Grundlage für klinische Anwendungen dar. Die Anwendung von Basen-Editing bei HD ist jedoch mit Herausforderungen verbunden.
Die Effektivität der Methode hängt von mehreren Faktoren ab, darunter die Dauer und Effizienz der Virustransduktion, das Risiko unerwünschter Off-Target-Editing-Effekte und die Verteilung der Therapie in den betroffenen Gehirnregionen. Zwar zeigen Studien in Mäusen, dass Off-Target-Effekte hauptsächlich in nicht-kodierenden oder synonymen Regionen auftreten und selten zu schädlichen Proteinveränderungen führen, doch sind Langzeitstudien zur Sicherheit und Wirksamkeit noch ausstehend. Zudem sind die neurodegenerativen Prozessen der HD komplex und multiparametrisch. Die Reduktion somatischer CAG-Expansionen sollte idealerweise mit weiteren therapeutischen Ansätzen kombiniert werden, wie etwa der Unterstützung der zellulären DNA-Reparaturmechanismen oder neuroprotektiven Therapien. Nichtsdestotrotz bietet die Basenbearbeitung einen neuartigen und spezifischen Zugang, der die molekulare Ursache von HD direkt adressiert.
Derzeit laufen Forschungsarbeiten, um die Methodik für den Humanbereich zu optimieren. Dazu zählt die Entwicklung noch effizienterer und präziserer Basen-Editoren, die auf bestimmte Zelltypen zielgerichtet agieren, die Verfeinerung der Virusvektoren für verbesserte Gewebepenetration sowie Studien zur bestmöglichen Zeit und Dosierung der Therapie. Insbesondere die Überwindung der Blut-Hirn-Schranke stellt weiterhin eine Herausforderung für die Verabreichung dar. Parallel zu HD wird das Basen-Editing auch bei anderen TNR-Erkrankungen, wie der Friedreich-Ataxie mit ihren GAA-Repeat-Expansionen, erfolgreich erforscht, was die universelle Bedeutung dieser Technologie unterstreicht. Die methodische Parallele beruht darauf, dass bei beiden Krankheiten Unterbrechungen die genomische Stabilität verbessern und somit den Krankheitsverlauf abmildern können.
Zusammenfassend lässt sich sagen, dass die Gen-Basenbearbeitung eine bahnbrechende Strategie darstellt, um die somatische Instabilität von CAG-Repeats bei Huntington zu reduzieren. Die Verringerung oder gar Umkehr der somatischen Expansion begrenzt die schädliche Ausdehnung der Polyglutamin-Kette im Huntingtin-Protein, womit ein direkter Einfluss auf Krankheitsprogression und Symptomatik möglich ist. Während noch mehrere Hürden vor klinischer Anwendbarkeit überwunden werden müssen, signalisieren aktuelle Studien enorme Fortschritte und eröffnen neue Wege zur Behandlung dieser bislang unheilbaren Erkrankung. Die Fokussierung auf somatische Wiederholungserweiterungen als therapeutisches Ziel könnte in den kommenden Jahren die Entwicklung von Heilmitteln entscheidend vorantreiben und das Leben zahlreicher Patienten weltweit nachhaltig verbessern. Die Kombination von präziser Gen-Editierung mit etablierten symptomatischen Therapien verspricht ein umfassendes Behandlungskonzept, das über die bisherige reine Symptomkontrolle hinausgeht.
Für Patienten und Angehörige bedeutet dies Hoffnung auf eine Zukunft, in der Huntington und ähnliche genetische Erkrankungen effizienter bekämpft werden können, indem direkt an den Wurzeln der genetischen Instabilität angesetzt wird. Wissenschaftler, Kliniker und die pharmazeutische Industrie sind gefordert, diese vielversprechenden Ansätze weiterzuentwickeln und sicher in die klinische Praxis zu integrieren.