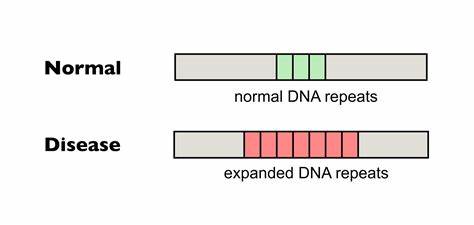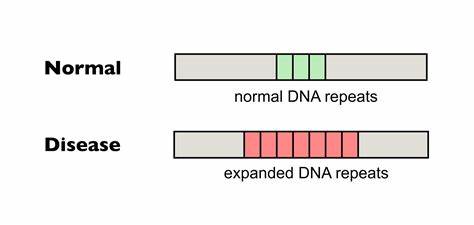
Die Huntington-Krankheit (HD) ist eine verheerende neurodegenerative Erkrankung, die durch eine Expansion von Trinukleotid-Wiederholungen, genauer gesagt CAG-Repeats, im HTT-Gen verursacht wird. Diese krankhaften Wiederholungen führen zu einer abnorm verlängerten Polyglutamin-Sequenz im Huntingtin-Protein, was schlussendlich zum Verlust von Nervenzellen, vor allem im Striatum des Gehirns, führt. Ein besonderes Merkmal dieser Wiederholungen ist ihre instabile Natur: Sie können mit der Zeit in bestimmten Geweben weiter verlängert werden, ein Phänomen, das als somatische Repeat-Expansion bezeichnet wird und maßgeblich die Ausprägung und den Verlauf der Erkrankung beeinflusst. Die Suche nach Strategien zur Stabilisierung oder Reduzierung dieser somatischen Expansionen ist damit von zentraler Bedeutung für die Entwicklung neuartiger Therapien bei HD. Die Arbeit im Bereich der Genom-Editierung hat in den letzten Jahren eine Revolution erfahren.
Insbesondere die sogenannten Base-Editing-Technologien bieten die Möglichkeit, gezielte Einzelbasen-Substitutionen ohne Einführung von Doppelstrangbrüchen durchzuführen. Neueste Studien zeigen, dass es möglich ist, die gefährlichen CAG-Wiederholungen gezielt zu bearbeiten und dabei sogenannte Unterbrechungen einzuführen, die natürlicherweise in einigen Populationen vorkommen und mit einer geringeren Instabilität der Wiederholungen assoziiert sind. Diese Interruptionssequenzen, meist durch die Veränderung von CAG zu CAA, kodieren zwar ebenfalls für Glutamin, verhindern jedoch die Bildung von sekundären DNA-Strukturen, die die Expansion fördern. Forschungsergebnisse demonstrieren, dass die gezielte Einführung solcher Unterbrechungen durch Cytosin-Base-Editoren (CBEs) in Humanzellen, einschließlich Fibroblasten von Patienten mit Huntington, sowohl effizient als auch sicher möglich ist. Die wiederholte Anwendung dieser Methode führte zu einer bedeutenden Reduktion der somatischen Expansionen in vitro, was auf eine Stabilisierung der pathogenen Repeat-Sequenz hinweist.
Weiterhin zeigten Tiermodelle mit humanisiertem HTT-Gen, die mit Adeno-assoziierten Viren (AAVs) die entsprechende Editierungsenzyme erhalten hatten, eine signifikante Verringerung der somatischen Repeat-Expansionen im Zentralnervensystem. Besonders bemerkenswert war dabei, dass diese Veränderungen nicht nur eine Verhinderung der Expansion bewirkten, sondern auch eine Verkürzung der bereits stark verlängerten Repeat-Sequenzen zur Folge hatten. Die Bedeutung dieser Befunde kann nicht hoch genug eingeschätzt werden. Die Länge der CAG-Wiederholungen bei Geburt bestimmt das Erkrankungsalter und die Schwere der HD, doch die somatische Expansion modifiziert den Verlauf dramatisch. Die Möglichkeit, diese somatische Instabilität zu limitieren oder gar rückgängig zu machen, bietet somit die Aussicht auf eine symptomverzögernde oder -vermindernde Therapie, die direkt an der molekularen Ursache ansetzt.
Darüber hinaus ist die Zielgenauigkeit dieser Base-Editing-Technologie hoch, wobei die Nebenwirkungen und Off-Target-Effekte in kontrollierten Studien minimiert werden konnten. Die meisten unerwünschten Editierungen betrafen nicht-kodierende oder synonyme Veränderungen, die wahrscheinlich keine negativen funktionellen Folgen tragen. Neben der Huntington-Krankheit steht auch die Friedreich-Ataxie (FRDA), eine erblich bedingte ataktische Erkrankung, im Zentrum ähnlicher Forschungsbemühungen. FRDA wird durch eine Expansion von GAA-Repeats im FXN-Gen verursacht, die zur Transkriptionshemmung und einem Mangel des mitochondrialen Proteins Frataxin führen. Analog zu den Beobachtungen bei HD bewirken natürliche Interruptionssequenzen in den GAA-Repeats eine Stabilisierung und mildern Krankheitsverlauf.
Beratung und Tests von Adenin-Base-Editoren (ABEs) zeigten eine effiziente Einführung eben solcher Interruptionssequenzen in patienteneigene Fibroblasten und in Modelltierlinien. Dies führte sowohl zu einer Reduktion der somatischen Expansion als auch zur Steigerung der FXN-Expression, was wichtige molekulare Korrelate einer potentiellen Therapie darstellt. Die Entwicklung von viralen Vektoren, speziell der AAV9-Serotypen, hat den Weg für effiziente In-vivo-Deliverierung dieser Editierungs-Tools gelegt. Neonatale intracerebroventrikuläre Injektionen ermöglichen die Zielansteuerung neuraler Zellpopulationen, die von den Repeat-Expansionen betroffen sind. Modelle mit analogen Somatischen Expansionen zeigen eine signifikante und nachhaltige Verringerung der Repeat-Längen nach Behandlung mit Base-Editoren.
Dies unterstützt den therapeutischen Ansatz, die Genome dieser Zellen nachhaltig zu „korrigieren“. Auf regulatorischer und therapeutischer Ebene eröffnen diese Fortschritte neue Horizonte. Die Präzision und geringe Toxizität des Base-Editings könnten die Hürden für klinische Anwendungen senken. Dennoch bleibt die umfassende Analyse möglicher Off-Target-Effekte und eine Evaluation der Langzeitsicherheit essenziell. Insbesondere die Auswirkungen seltener Protein-verändernder Mutationen durch Off-Target-Editing müssen sorgfältig überwacht und bewertet werden, um potenzielle Risiken abschätzen zu können.
Die Fortschritte bei der Bearbeitung von Trinukleotid-Wiederholungen stellen einen Paradigmenwechsel in der Behandlung dominanter neurodegenerativer Erkrankungen wie Huntington und Friedreich-Ataxie dar. Durch die gezielte Einführung natürlicher Interruptionssequenzen wird die genomische Instabilität reduziert, die krankheitsbegünstigende somatische Expansion behindert und die pathologische Proteinexpression modifiziert. Diese molekularen Eingriffe schlagen eine Brücke von der Grundlagenforschung hin zu möglichen klinischen Interventionen. Die technologischen Grundlagen, bestehend aus verbesserten Base-Editoren wie CBE4max und ABE8e sowie der optimierten sgRNA-Strategie, ermöglichen eine zunehmend präzise Navigation durch repetitive Genomelemente. Grundlage hierfür ist auch die sorgfältige Evaluierung in verschiedenen Modellssystemen – von humanen Zellkulturen bis zu „humanisierten“ Mausmodellen –, die die Übertragbarkeit und Effektivität der Methode belegen.
Zusammenfassend lässt sich festhalten, dass die gezielte Bearbeitung pathogener Genregionen bei Trinukleotid-Repeat-Erkrankungen einen wichtigen Türöffner für personalisierte, krankheitsmodifizierende Therapien darstellt. Die Reduktion der somatischen Repeat-Expansionen in Patientenzellen durch Base Editing bringt Hoffnung auf eine zukünftige Behandlung, die nicht nur die Symptome mildert, sondern die Ursache auf DNA-Ebene adressiert und somit das Fortschreiten der Erkrankung entscheidend verlangsamen oder stoppen kann. Die weitere Forschung wird entscheidend sein, um optimale Editierungsstrategien zu entwickeln, Sicherheitsaspekte zu klären und den Übergang in klinische Studien zu realisieren.