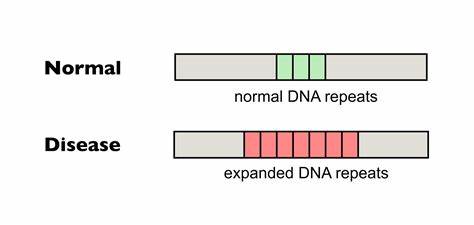
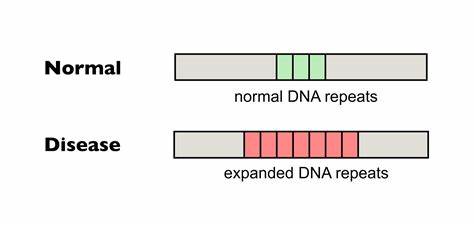
Die Huntington-Krankheit (HD) zählt zu den sogenannten trinukleotid-Wiederholungserkrankungen und stellt Betroffene sowie Forschende vor enorme Herausforderungen. Ursache der Erkrankung ist eine pathologische Expansion von CAG-Trinukleotid-Wiederholungen im HTT-Gen, die zu einer fehlerhaften Bildung des Huntingtin-Proteins führt. Die Länge dieser repetitiven Sequenz ist essenziell für den Krankheitsverlauf: Je größer die Wiederholungszahl, desto früher beginnt die Erkrankung in der Regel, und desto schwerer gestaltet sich deren Verlauf. Doch neben der erblichen Konstitution sind vor allem sogenannte somatische Repeat-Expansionen in bestimmten Körperzellen von großer Bedeutung, denn sie verstärken die Schädigung im Laufe des Lebens – ein Prozess, den Forscherinnen und Forscher jetzt mit Hilfe von modernstem Genom-Editing effektiv zu reduzieren versuchen. Somatische Repeat-Expansionen beschreiben eine zelluläre Instabilität, bei der die Anzahl der CAG-Wiederholungen in den betroffenen Zellen über die ursprüngliche Keimbahnmutation hinaus anwächst.
Besonders in Neuronen des Zentralnervensystems führt dies zu einer verstärkten Ansammlung toxischer Proteinfragmente, die letztlich zum Absterben der Zellen beiträgt und sich in den charakteristischen Symptomen wie motorischer Dysfunktion, kognitiven Einbußen und psychiatrischen Problemen manifestiert. Bisher war eine direkte Therapie, die auf die Stabilisierung oder Korrektur der DNA-Wiederholungen abzielt, jedoch nicht verfügbar. Ein vielversprechender Ansatz basiert auf sogenannten Basen-Editoren, einer innovativen Klasse von Genome-Editing-Tools, die gezielt einzelne Nukleotide in der DNA austauschen können, ohne dabei Doppelstrangbrüche zu verursachen. Im Gegensatz zu klassischen CRISPR-Cas9-Systemen schneiden diese Enzyme die DNA nicht vollständig durch, sondern verändern einzelne Basen in einer kontrollierten Weise. Das kann genutzt werden, um die tödlichen CAG-Reihenfolgen im Huntingtin-Gen gezielt und präzise anzureichern mit sogenannten Unterbrechungen – Sequenzvarianten, die natürlicherweise in gesünderen Allelen vorkommen und mit einer geringeren Instabilität sowie einem verzögerten Krankheitsbeginn assoziiert sind.
Aktuelle Studien zeigen, dass die Einführung dieser Interruptions in Patientenzellen direkte Auswirkungen auf die Stabilität der CAG-Wiederholungen hat. Durch die gezielten Cytosin- und Adenin-Basen-Editoren lassen sich CAG-Wiederholungen in CAA-Trinukleotide umwandeln – eine stille Mutation, die zwar die Aminosäuresequenz des Proteins unberührt lässt, aber die Neigung zur weiteren Expansion der repetitiven Regionen deutlich vermindert. Untersuchungen an humanen Fibroblasten von Huntington-Patienten belegen, dass nach Behandlung mit solchen Basen-Editoren die somatische Expansion von CAG-Repeats reduziert und teilweise sogar eine Kontraktion der Wiederholungen erreicht werden kann. Die Umsetzung dieser molekularen Strategien in lebende Organismen wurde anhand von genetisch veränderten Mäusen demonstriert, die das menschliche HTT-Gen mit pathologisch langen CAG-Repeats tragen. Die Injektion von viralen Vektoren, die die Basen-Editoren transportieren, in das zentrale Nervensystem von neugeborenen Mäusen, führte zu einer stabilen und spezifischen Integration von CAA-Unterbrechungen in den pathogenen CAG-Tracts.
Die Folge war eine signifikante Verringerung der somatischen Repeatexpansionen im Gehirn dieser Tiere, insbesondere in relevanten Hirnregionen wie dem Striatum und der Großhirnrinde. Bemerkenswert ist, dass sich die therapeutische Wirkung über Wochen und Monate aufrechterhielt, was für eine anhaltende Stabilisierung der genomischen Sequenzen spricht. Neben dem Nutzen für das Wesen der Erkrankung weist diese Behandlungsmethode auch eine bemerkenswerte Spezifität auf. Das gezielte Editieren der CAG-Repeats führt zu sehr wenigen Off-Target-Effekten, also unerwünschten genetischen Veränderungen an anderen Stellen im Genom. Um die Sicherheit der Methode besser einschätzen zu können, wurden umfassende Analysen durchgeführt, die zeigen, dass ein Großteil der potenziellen Off-Target-Loci entweder nicht bearbeitet wurde oder nur neutrale Veränderungen aufwies.
Dort, wo aminoacidverändernde Mutationen auftraten, handelte es sich meist um harmlosere Varianten. Dennoch sind weitere Langzeitstudien sinnvoll, um mögliche Nebenwirkungen auszuschließen und die Translatierbarkeit in klinische Anwendungen zu ermöglichen. Wichtig für die zukünftige Therapieentwicklung ist auch der Erfolg bei der Optimierung der Basen-Editoren selbst. Verschiedene Varianten der Enzyme und Trägerproteine wurden getestet, um die höchste Effizienz bei gleichzeitig minimaler Toxizität zu erreichen. So konnten durch die Verwendung von Cas9-Varianten mit erweiterten PAM-Erkennungssequenzen mehr Zielstellen erreicht und eine größere Anzahl von Wiederholungen editiert werden.
Auch die Dosierung und der zeitliche Verlauf der Behandlung erwiesen sich als entscheidend für den maximalen Erfolg. Auf gesellschaftlicher Ebene eröffnet das Editieren der CAG-Wiederholungen neue Perspektiven für die Lebensqualität der Patienten. Dauerhafte Veränderungen auf DNA-Ebene versprechen eine nachhaltige Wirkung bereits nach einmaliger Behandlung, was sich deutlich von den meisten symptomatischen Therapien unterscheidet, die auf kurzfristige Entlastung ausgelegt sind. Zudem könnte durch frühe Intervention, etwa im Kindesalter, das Fortschreiten der Krankheit signifikant verlangsamt oder sogar verhindert werden. Dennoch existieren noch Hürden bei der klinischen Umsetzung.
Die effiziente und sichere Abgabe von Basen-Editoren in menschliche Gehirnregionen bleibt eine Herausforderung. Die verwendeten viralen Vektoren weisen zwar eine gute Neuropatienten-Tropismus auf, die Immunantwort und mögliche Langzeitwirkungen müssen jedoch weiterhin genau beobachtet werden. Weiterhin sind neben neuronalen auch andere Zelltypen bei Huntington beteiligt, weshalb ein umfassendes Therapieschema noch konzipiert werden muss. Insgesamt zeigen die bahnbrechenden Fortschritte im Bereich der Genom-Editierung, dass das direkte Verändern von DNA-Sequenzen zur Stabilisierung von Tandem-Wiederholungen bei Huntington-Patienten effektiv möglich ist. Diese Innovationen markieren einen Paradigmenwechsel im Umgang mit erblichen neurologischen Erkrankungen und weisen den Weg zu personalisierten und kurativen Therapien.
Mit weiteren Forschungen und klinischen Studien könnte diese Methode künftig Teil der Behandlung von Huntington und anderen TNR-bedingten Erkrankungen werden und den Betroffenen neue Hoffnung schenken.




![Domain/OS Design Principles (1989) [pdf]](/images/1E5E6EAD-D0D8-4814-A356-4FED55AA17AE)

