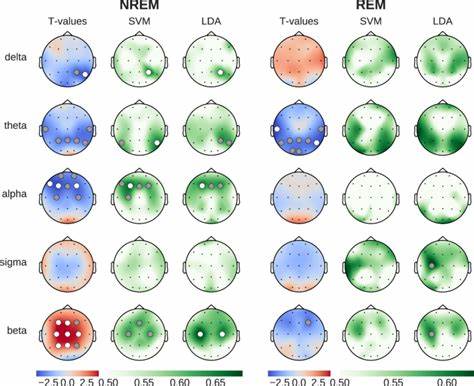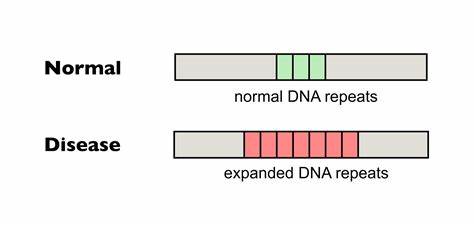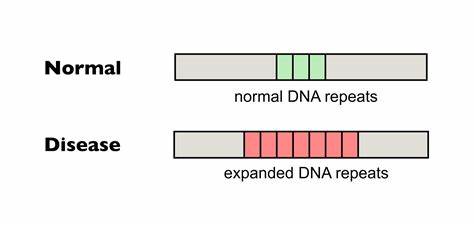
Die Huntington-Krankheit (HD) stellt eine ernsthafte neurodegenerative Erkrankung dar, die durch die Expansion von CAG-Trinukleotid-Wiederholungen im Huntingtin-Gen (HTT) verursacht wird. Diese abnormale Wiederholung führt zur Produktion eines toxischen Proteins, das im Gehirn Nervenzellen schädigt und nach fortschreitendem Verlauf zum Verlust motorischer und kognitiver Fähigkeiten führt. In den letzten Jahren rückte ein ebenfalls bedeutsamer Mechanismus zunehmend in den Fokus der Forschung: die somatische Expansion dieser CAG-Wiederholungen und deren Einfluss auf den Krankheitsverlauf. Neue Erkenntnisse und Technologien ermöglichen heute die gezielte Bearbeitung dieser Wiederholungen, was das Fortschreiten der Krankheit potenziell abbremsen oder gar verhindern kann. Bei Huntington-Trios handelt es sich um fortlaufende Wiederholungen der Nukleotidsequenz CAG in bestimmten Genabschnitten.
Diese Sequenz kodiert für die Aminosäure Glutamin, weshalb man von sogenannten Polyglutamin-Krankheiten spricht. Von besonderer Bedeutung ist dabei nicht nur die Länge der Wiederholung bei Geburt, sondern auch deren Veränderung im Laufe des Lebens, speziell im zentralen Nervensystem (ZNS). Es wurde festgestellt, dass die somatische Instabilität — das heißt die Neigung der CAG-Repeats, sich in einzelnen Körperzellen weiter zu verlängern — maßgeblich mit der Schwere der Symptome und dem Alter bei Krankheitsbeginn korreliert. Diese Expansionen sind komplexen molekularen Mechanismen unterworfen. Fehlerhafte DNA-Reparatursysteme, die während der Zellteilung oder Transkription entstehen, fördern insbesondere bei längeren CAG-Strecken eine Verzögerung oder gar eine Expansion der repetitiven Sequenzen.
Die Folge sind toxische Veränderungen in den betroffenen Neuronen, die letztlich den neurodegenerativen Prozess beschleunigen. Daher ist die Hemmung oder Reduktion dieser somatischen Expansionen ein vielversprechender therapeutischer Ansatz. Eine natürliche Variante, die in der Bevölkerung beobachtet wird, betrifft sogenannte Interruptions oder Unterbrechungen im CAG-Repeat. Diese bestehen oft aus CAA-Sequenzen, die zwar ebenfalls Glutamin kodieren, aber die Wiederholung aufbrechen und dadurch die Bildung destabiler DNA-Strukturen verhindern. Studien belegen, dass Menschen mit solchen Interruptions eine verzögerte Onset der Huntington-Symptome aufweisen und insgesamt mildere Verlaufsformen erlebt haben.
Das zeigt schon heute, wie wichtig die Struktur der Wiederholung ist. Basierend auf diesen Erkenntnissen entwickelte sich die Idee, mithilfe modernster Gen-Editierungstechnologien, insbesondere der sogenannten Basen-Editierung (Base Editing), gezielt diese Interruptions zu erzeugen. Im Gegensatz zu klassischen CRISPR/Cas9-Technologien müssen dabei keine Doppelstrangbrüche in der DNA erzeugt werden. Vielmehr werden einzelne Basen umgewandelt, was präziser und nebenwirkungsärmer ist. Forscher stellten verschiedene Cytosin- und Adenin-Baseneditoren her, die in der Lage sind, bestimmte Nukleotide in den CAG- oder GAA-Trinukleotid-Regionen umzuwandeln.
Dadurch entstehen Interruptions wie CAA in CAG-Repeats oder GGA/GAG in GAA-Repeats, welche den somatischen Instabilitäten entgegenwirken. Die Herausforderung dabei liegt in der hohen Wiederholungshäufigkeit, der möglichen Off-Target-Effekten und der stabilen Ausbringung der Editoren in die betroffenen Nervenzellen. In Laborstudien und Zellmodellen konnte gezeigt werden, dass diese gezielte Bearbeitung von CAG-Repeats in patienteneigenen Fibroblasten zu einer signifikanten Reduktion der somatischen Expansion führt. Die Anzahl der Interruptions erhöhte sich nach der Behandlung, und es zeigte sich eine Tendenz zu kürzeren, stabileren Repeat-Längen über längere Kultivierungszeiten. Dies lässt darauf schließen, dass die eingeschleusten Interruptions tatsächlich die instabile Verlängerung der Wiederholungen hemmen können.
Darüber hinaus gelang es durch den Einsatz von Adeno-assoziierten Viren (AAV9) in Mausmodellen der Huntington-Krankheit, die Baseneditoren effizient in das Gehirn zu bringen. Insbesondere im Kortex und Striatum, den Hauptbereichen, die bei HD betroffen sind, konnte eine substanzielle Anzahl von HTT-Allelen mit Interruptions erzeugt werden. Über Wochen und Monate führte dies zu einer signifikanten Verminderung der somatischen CAG-Expansion in vivo. Bemerkenswert ist, dass neben einer Prävention der Expansion auch eine leichte Kontraktion, also Verkürzung der Wiederholungen, beobachtet wurde, was für den potenziellen Therapieerfolg von großem Vorteil sein könnte. Parallel dazu wurde ein ähnliches Vorgehen bei Friedreich-Ataxie (FRDA) getestet, einer anderen TNR-bedingten neurodegenerativen Erkrankung, ausgelöst durch GAA-Repeat-Expansionen im FXN-Gen.
Die Einführung von Interruptions durch Adeninbasen-Editierung konnte in patientenabgeleiteten Zellen die Transkription des betroffenen Gens erhöhen und die Somatikin-Stabilität verbessern. Auch hier wurden in Mausmodellen signifikant weniger schädliche somatische Repeat-Expansionen nachgewiesen. Die Evaluierung möglicher Nebenwirkungen und Off-Target-Effekte ist dabei ein zentrales Anliegen der Forschung. Analysen auf Genomebene mittels umfangreicher Sequenzierungstechniken zeigten, dass die meisten unerwünschten Veränderungen entweder in nicht-kodierenden Bereichen der DNA auftreten oder zu Synonymen Mutationen führen, die keine Änderung der Proteinstruktur bewirken. Ein kleiner Anteil an potenziell funktionell relevanten Mutationen erfordert jedoch noch weitergehende Studien hinsichtlich Langzeitfolgen, insbesondere in zentralnervösen Zelltypen.
Ein weiterer stets diskutierter Punkt betrifft die Verabreichung der Gen-Editierungssysteme. Die Verwendung von AAV9-Vektoren in frühen Entwicklungsstadien, beispielsweise durch neonatalen intraventrikulären (ICV) Injektionen, sorgte für eine effiziente Transduktion von Nervenzellen im Mausmodell. Zugleich ist es wünschenswert, alternative Serotypen oder Verabreichungswege zu untersuchen, die eine bessere Ausbreitung im ZNS oder auch den Targeting von Nicht-Neuronen, wie Gliazellen oder Herzmuskelzellen, gewährleisten. Denn besonders bei FRDA ist das Herz betroffen, was eine umfassendere Therapie nötig macht. Diese bahnbrechenden Studien eröffnen neue Perspektiven für therapeutische Interventionen bei Huntington und anderen TNR-Erkrankungen.
Die Basen-Editierung zur Erzeugung von Interruptions stellt einen eleganten Weg dar, um die molekulare Basis der Erkrankung präzise zu verändern. Indem somatische Expansionen der pathogenen Wiederholungen reduziert und sogar leichte Kontraktionen induziert werden, lässt sich die Progression der neurodegenerativen Prozesse theoretisch verzögern oder aufhalten. Zukunftsforschungen werden sich darauf konzentrieren, die Sicherheit, Spezifität und Effizienz dieser Technologien weiter zu verbessern. Optimierungen der Botenstoffe oder viralen Vektoren zur Verabreichung, schnellere und gezieltere Kontrollmechanismen zur Minimierung von Off-Target-Effekten sowie genauere Untersuchungen zu den Auswirkungen in neuronalen und nicht-neuronalen Zelltypen sind essenziell. Darüber hinaus sollte die Wirkung der somatischen Repeat-Interruptionen auf das klinische Bild der Patienten erforscht werden.
Obwohl die Modelle vor allem molekulare und genetische Effekte zeigen, will man bald auch den Einfluss auf motorische Fähigkeiten, Kognition und Lebensqualität erfassen. Die zunehmende Genauigkeit der präklinischen Modelle wird helfen, diese Fragen zu klären und weiterführende personalisierte Therapieansätze zu ermöglichen. Nicht zuletzt unterstreicht die Arbeit auch die Bedeutung der natürlichen genetischen Variation bei TNR-Erkrankungen. Mittels Genomanalysen wird immer deutlicher, dass einzelne natürlichen Unterbrechungen oder Variationen der Wiederholungen großen Einfluss auf Krankheitsschwere und Progression haben. Dies nicht nur für Huntington, sondern auch im Bereich von Spinocerebellären Ataxien, Fragilem X-Syndrom und anderen seltenen Erkrankungen.
Insgesamt sind die Fortschritte in der Bearbeitung der trinukleotidalen Repeat-Längen ein überzeugender Beleg dafür, dass präzise genomische Veränderungen therapeutisch nutzbar gemacht werden können, um komplexe neurodegenerative Erkrankungen wie Huntington gezielt an der molekularen Ursache anzugehen. Die Kombination aus genetischem Verständnis, innovativer Basen-Editierung und moderner Gentherapie bietet neue Hoffnung für eine bislang unheilbare Krankheit und öffnet Türen zu einer neuen Ära der personalisierten Medizin.