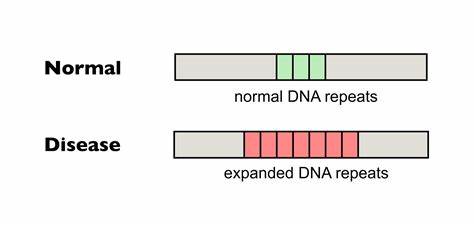
Die Huntington-Krankheit (HD) gehört zu den sogenannten trinukleotid-Repeat-Erkrankungen, die durch abnorm lange Wiederholungen eines dreibasigen DNA-Sequenzabschnitts entstehen. Im Fall von HD handelt es sich um die pathologische Expansion der CAG-Trinukleotidsequenz im HTT-Gen, die zur Produktion eines fehlerhaften Huntingtin-Proteins führt und schädliche neurodegenerative Prozesse im Gehirn auslöst. Bisher existieren keine zugelassenen Therapien, die den Verlauf der Krankheit aufhalten oder rückgängig machen können, weshalb die Entwicklung innovativer molekularer Strategien von höchster Bedeutung ist. Eine zentrale Ursache für die Schädigung bei Huntington’s ist die somatische Expansion dieser CAG-Wiederholungen, also die weitere Verlängerung der Repeat-Segmente in bestimmten Körperzellen über die Lebensspanne hinweg. Je länger die CAG-Stränge werden, desto höher ist die Wahrscheinlichkeit für das Auftreten von Krankheitssymptomen sowie für deren Schweregrad und frühes Auftreten.
Die Zellen im zentralen Nervensystem, besonders die Neuronen in der Striatumregion, sind besonders anfällig für diese Instabilitäten, welche letztlich zu einem beschleunigten neurodegenerativen Zerfall führen. Der Meilenstein in der aktuellen Forschung ist die Anwendung sogenannter Base-Editing-Verfahren, die eine präzise Änderung einzelner DNA-Basen ohne Doppelstrangbrüche ermöglichen. Diese Technologie erlaubt es, gezielt einzelne Nukleotide in den CAG-Repeatsequenzen zu verändern, um diese vor weiterer Expansion zu schützen. Anstatt die Anzahl der Wiederholungen zu verringern, geht die Strategie hier einen ungewöhnlichen Weg: Durch Einfügung von nicht-pathogenen Unterbrechungen in die CAG-Repeats, zum Beispiel der CAA-Sequenz, wird die DNA-Struktur so stabilisiert, dass ein weiterer Längenanstieg erschwert wird. In Patientenfibroblasten und in Zellkulturmodellen gelang es mithilfe von Cytosin-Base-Editoren (CBEs), die CAG-Wiederholungen gezielt zu unterbrechen.
Durch das Anbringen von einzelnen CAA-Codons in den langgezogenen CAG-Strecken konnte die somatische Expansion in vitro klar vermindert werden. Bemerkenswert ist, dass diese Unterbrechungen die Basensequenz zwar verändern, aber aufgrund der Synonymität derselben Aminosäure – Glutamin – keine Veränderung des Huntingtin-Proteins bewirken, sodass die Proteinfunktion unangetastet bleibt. In Mausmodellen, die ein humanisiertes HTT-Gen mit toxisch langen CAG-Tracks tragen, wurde die Base-Editing-Technologie mittels AAV9-Vektoren in das zentrale Nervensystem eingebracht. Die adeno-assoziierten Viren ermöglichen eine neuronenspezifische und effiziente Transduktion vor allem im Kortex und Striatum, den zentralen Hirnregionen, die von HD betroffen sind. Die Studienergebnisse zeigten eine effektive Conversion von mehreren CAGs in CAA innerhalb der Repeat-Trakte, die somatische Ausdehnung der Pathogenese war im Vergleich zu unbehandelten Tieren signifikant reduziert, und man beobachtete sogar erste Anzeichen von Kontraktion der Repeatlängen.
Die Bedeutung dieser Ergebnisse liegt nicht nur im direkten Behandlungspotential, sondern auch darin, dass sie die bisherige Annahme stärken, dass die Länge von reinen, ununterbrochenen CAG-Strecken die treibende Kraft hinter der Instabilität und dem Krankheitsausbruch ist. Die Einfügung von Unterbrechungen, die bei einigen Menschen natürlich vorkommen und eine mildere Krankheitsausprägung bedingen, kann somit gezielt im Genom nachgebildet werden, um therapeutische Effekte zu erzeugen. Neben Huntington’s wurde die gleiche Base-Editing-Technik auch auf den GAA-Repeat im FXN-Gen angewandt, dessen Expansion Friedreich-Ataxie verursacht. Dort fungiert ein ähnliches Prinzip: Adenin-Base-Editoren (ABEs) bringen nicht-pathogene Unterbrechungen in den langen GAA-Repeats ein und schaffen so eine stabilere Genomstruktur. In fibroblastären Patienten-Zelllinien und in entsprechenden Mausmodellen führte diese Modifikation zu einer signifikanten Abnahme der somatischen Repeatexpansion.
Zudem wurde eine teilweise Wiederherstellung der FXN-Genexpression beobachtet, was eine wichtige molekulare Verbesserung für die Krankheit darstellt. Ein zentrales Thema der Forschung ist die Präzision und Sicherheit des Base-Editings, denn die eingesetzten Guide-RNAs (sgRNAs), die die Editors an die richtigen DNA-Stellen lenken, könnten theoretisch auch ähnliche Repeat-Sequenzen an anderen Genorten bearbeiten. Um diese potenziellen Off-Target-Effekte zu analysieren, wurde eine Kombination aus genomweiten Sequenzmethoden wie CIRCLE-seq und hochauflösender Whole-Genome-Sequenzierung eingesetzt. Dabei zeigte sich, dass die meisten Off-Target-Bearbeitungen in nicht-kodierenden Regionen liegen oder sehr seltene, oft synonyme Veränderungen in Proteincodierenden Bereichen bewirken. Die Induktion von pathogenen Mutationstypen war äußerst gering, und viele Veränderungen wurden als ähnliche genetische Varianten erkannt, die auch in der menschlichen Population vorkommen.
Die Langzeitwirkung eines einzelnen AAV-gestützten Base-Editor-Transgens wurde in den Mausmodellen über Wochen bis Monate beobachtet. Die kontinuierliche Expression und Aktivität des Editors führte zu einem zunehmenden Anteil editierter Allele und damit einer kumulativen Verfestigung der unterbrochenen Repeat-Trakte. Diese Stabilisierung verringert nicht nur die Ausdehnung, sondern fördert auch die Kontraktion schon pathologisch langer Repeat-Trakte, was in Zukunft besonders wichtig sein könnte, um betroffene Patienten über einen therapeutischen Zeitrahmen hinweg zu behandeln. Nicht zuletzt verstärkt die Methode die Hoffnung auf eine klinisch relevante Intervention für Huntington’s und verwandte Repeat-Expansion-Erkrankungen. Derzeitige Therapien adressieren meist die Symptome oder versuchen, die Expression des mutierten Proteins zu reduzieren.
Die präzise Genomkorrektur hingegen verändert die genetische Ursache, ohne dabei das normale Protein oder die Genfunktion zu beeinträchtigen, was eine nachhaltige, krankheitshemmende Wirkung verspricht. Trotz all dieser Erfolge stehen Herausforderungen für die klinische Anwendung an. Effizienz und Spezifität des Editor-Systems müssen weiter optimiert werden, um potenzielle Nebenwirkungen zu minimieren. Auch die gezielte Verteilung des Genom-Editors in verschiedenen betroffenen Geweben, wie z. B.
in nicht-neuronalen Zellen bei Friedreich-Ataxie, muss adressiert werden. Die Entwicklung alternativer Vektoren und Verabreichungswege könnte hier Verbesserungen bringen. Zusammenfassend bietet die Base-Editing-Technologie eine revolutionäre Perspektive für die Behandlung der Huntington-Krankheit und anderer Trinukleotid-Repeat-Erkrankungen. Durch die gezielte Strukturmodifikation pathologischer DNA-Repeats wird das Fortschreiten der Krankheit auf molekularer Ebene gebremst. Die Erforschung von Off-Target-Effekten und Langzeitfolgen wird zurzeit intensiv vorangetrieben, um die Sicherheit für zukünftige Therapien zu gewährleisten.
Die bisherigen Studien in Patienten-Zellkulturen und Tiermodellen sind vielversprechend und könnten in nicht ferner Zukunft den klinischen Standard nachhaltig verändern.